Molecular Docking – in silico drug designing
By Ankita Tah and Santanu Roy, Post Graduate Department of Microbiology, Acharya Prafulla Chandra College, New Barrackpore, Kolkata – 700 131.
Neucrad Health India, April 25, 2019
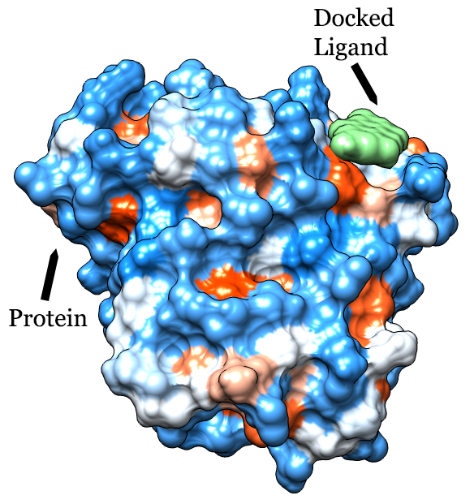
Molecular docking is an algorithmic process done in a computerized manner to find out the best binding site between two molecules, which include a target macromolecule, e.g., protein or nucleic acid, and a small molecule, such as a phytochemical that acts as ligand. Docking is one of the principal tools for in silico drug designing [1].
Initially, a target needs to be identified for functional modulation. Then appropriate ligands are identified against that target so that the ligands can modulate its function in such a way that it leads to some consequential effects as desired prior to the docking process. When a ligand binds to a target, the target undergoes a conformational change to find out the best fitting position of the ligand with the target. In order to function properly a ligand needs to bind to the target reversibly, otherwise most of the biological processes that involve the target may be potentially hindered [2]. Bioinformatics has made it is easy to locate the active sites in targets such as proteins and to calculate the energies of interactions between the targets and several types of ligands very quickly.
There are a number of protein-ligand docking programs. Some of these are freeware, some are free for academic use only and some are commercial in nature, needing subscription. Some of these are listed in the following table, along with their publishers, year of publishing, operating systems supported and types of license available.
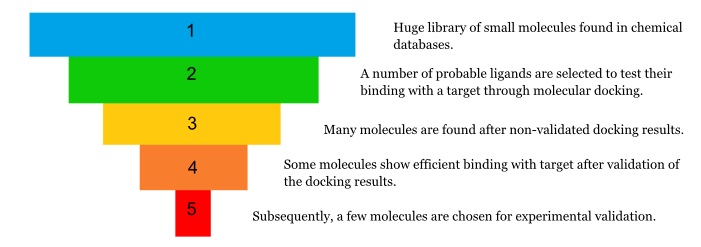
Applications of molecular docking include structure based drug designing. By comparing the affinity of small molecules, i.e., ligands, towards target protein(s), using the tools listed in the table, one can predict the best candidates to be treated as lead compounds in in vitro experiments. The main advantage of in silico drug designing lies in the fact that it is much more time saving than looking for probable drug candidates experimentally in the laboratory. A large number of potential drug targets can be screened in silico in a high throughput manner to quickly identify a handful of effective lead candidates which can then be tested experimentally.
Additionally, there are some software using which docking can be performed between a protein receptor and a small peptide. CABS-dock is such a web-based tool which is freely available, published by the University of Warsaw, which can be used for modeling protein-peptide interactions.
It is necessary to validate the Docking result to ensure the efficiency of the process. To validate the docking result there are several procedures. Some of them are mentioned below.
- One can validate the result by repeating the same docking procedure with the same target using different non-specific ligands. If the non-specific ligand binds within the same groove of the target where the specific ligand binds, it shows that the docking software used is not efficient to differentiate between specific and non-specific bindings.
- One can redo the docking of the same macromolecule and ligand pair but using different software. If the ligand binds in the same position into the target’s active site, it means the docking result is accurate.
References:
1. Naqvi, A.A.T., et al., Advancements in Docking and Molecular Dynamics Simulations Towards Ligand-receptor Interactions and Structure-function Relationships. Curr Top Med Chem, 2018. 18(20): p. 1755-1768.
2. Selzer, P.M., R.J. Marhofer, and A. Rohwer, Structure based rational drug design, in Applied bioinformatics. 2008, Springer. p. 126-127.
Let’s meet the authors

Ankita Tah
Post Graduate Department of Microbiology,
Acharya Prafulla Chandra College, New Barrackpore, Kolkata – 700 131.

Dr. Santanu Roy
Post Graduate Department of Microbiology,
Acharya Prafulla Chandra College, New Barrackpore, Kolkata – 700 131.